Database
PTID
-
 We present an integrated platform called Pesticide-Target interaction database (PTID), which comprises a total of 1347 pesticides with rich annotation of ecotoxicological and toxicological data as well as 13 738 interactions of pesticide-target and 4245 protein terms via text mining. Additionally, through the integration of ChemMapper, an in-house computational approach to polypharmacology, PTID can be used as a computational platform to identify pesticides targets and design novel agrochemical products.
We present an integrated platform called Pesticide-Target interaction database (PTID), which comprises a total of 1347 pesticides with rich annotation of ecotoxicological and toxicological data as well as 13 738 interactions of pesticide-target and 4245 protein terms via text mining. Additionally, through the integration of ChemMapper, an in-house computational approach to polypharmacology, PTID can be used as a computational platform to identify pesticides targets and design novel agrochemical products.
Web Server
iDrug
-
 iDrug is a versatile, user-friendly, and efficient online tool for computer-aided drug design based on pharmacophore and 3D molecular similarity searching. The web interface enables binding sites detection, virtual screening hits identification, and drug targets prediction in an interactive manner through a seamless interface to all adapted packages (e.g., Cavity, PocketV.2, PharmMapper, SHAFTS). Several commercially available compound databases for hit identification and a well-annotated pharmacophore database for drug targets prediction were integrated in iDrug as well. The web interface provides tools for real-time molecular building/editing, converting, displaying, and analyzing. All the customized configurations of the functional modules can be accessed through featured session files provided, which can be saved to the local disk and uploaded to resume or update the history work.
iDrug is a versatile, user-friendly, and efficient online tool for computer-aided drug design based on pharmacophore and 3D molecular similarity searching. The web interface enables binding sites detection, virtual screening hits identification, and drug targets prediction in an interactive manner through a seamless interface to all adapted packages (e.g., Cavity, PocketV.2, PharmMapper, SHAFTS). Several commercially available compound databases for hit identification and a well-annotated pharmacophore database for drug targets prediction were integrated in iDrug as well. The web interface provides tools for real-time molecular building/editing, converting, displaying, and analyzing. All the customized configurations of the functional modules can be accessed through featured session files provided, which can be saved to the local disk and uploaded to resume or update the history work. ProfKin
-
 ProfKin is a method developed for structure-based kinase profiling for small molecules of interest, which is established based on an in-house comprehensive structural database (KinLigDB) of manually curated kinase-ligand complex structures and associated information. Kinase profiling is an efficient strategy for kinase inhibitor discovery, polypharmacological drug discovery and drug repositioning. A variety of methods have been established for kinase profiling, of which the combined mode of experimental and computational approaches is attractive, particularly in the early drug discovery. With the increasing number of kinase-inhibitor complex structures, structure-based kinase profiling is of great interest.
ProfKin is a method developed for structure-based kinase profiling for small molecules of interest, which is established based on an in-house comprehensive structural database (KinLigDB) of manually curated kinase-ligand complex structures and associated information. Kinase profiling is an efficient strategy for kinase inhibitor discovery, polypharmacological drug discovery and drug repositioning. A variety of methods have been established for kinase profiling, of which the combined mode of experimental and computational approaches is attractive, particularly in the early drug discovery. With the increasing number of kinase-inhibitor complex structures, structure-based kinase profiling is of great interest. SiteMapper
-
 SiteMapper is a free web server for binding site comparison based on residue match and deviation minimization. Taking two user defined binding sites as reference and fitting structure, SiteMapper try to align these two structures in order to obtain a correspondence between residues from reference site and fitting site and evaluate the similarity between these two binding sites. SiteMapper can be very useful for evaluating the binding site similarity or performing discontinuous sequence alignment for binding sites.
SiteMapper is a free web server for binding site comparison based on residue match and deviation minimization. Taking two user defined binding sites as reference and fitting structure, SiteMapper try to align these two structures in order to obtain a correspondence between residues from reference site and fitting site and evaluate the similarity between these two binding sites. SiteMapper can be very useful for evaluating the binding site similarity or performing discontinuous sequence alignment for binding sites. PharmMapper
-
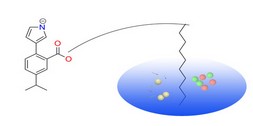 PharmMapper Server is a freely accessed web-server designed to identify potential target candidates for the given probe small molecules (drugs, natural products, or other newly discovered compounds with binding targets unidentified) using pharmacophore mapping approach. Benefited from the highly efficient and robust mapping method, PharmMapper bears high throughput ability and can identify the potential target candidates from the database within a few hours. PharmMapper is backed up by a large, in-house repertoire of pharmacophore database extracted from all the targets in TargetBank, DrugBank, BindingDB and PDTD. Over 7,000 receptor-based pharmacophore models (covering 1,627 drug targets information, 459 of which are human protein targets) are stored and accessed by PharmMapper.
PharmMapper Server is a freely accessed web-server designed to identify potential target candidates for the given probe small molecules (drugs, natural products, or other newly discovered compounds with binding targets unidentified) using pharmacophore mapping approach. Benefited from the highly efficient and robust mapping method, PharmMapper bears high throughput ability and can identify the potential target candidates from the database within a few hours. PharmMapper is backed up by a large, in-house repertoire of pharmacophore database extracted from all the targets in TargetBank, DrugBank, BindingDB and PDTD. Over 7,000 receptor-based pharmacophore models (covering 1,627 drug targets information, 459 of which are human protein targets) are stored and accessed by PharmMapper. ChemMapper
-
 ChemMapper is an online platform to predict polypharmacology effect and mode of action for small molecules based on 3D similarity computation. ChemMapper collects 4350 000 chemical structures with bioactivities and associated target annotations (as well as 43 000 000 non-annotated compounds for virtual screening). Taking the user-provided chemical structure as the query, the top most similar compounds in terms of 3D similarity are returned with associated pharmacology annotations. ChemMapper is designed to provide versatile services in a variety of chemogenomics, drug repurposing, polypharmacology, novel bioactive compounds identification and scaffold hopping studies.
ChemMapper is an online platform to predict polypharmacology effect and mode of action for small molecules based on 3D similarity computation. ChemMapper collects 4350 000 chemical structures with bioactivities and associated target annotations (as well as 43 000 000 non-annotated compounds for virtual screening). Taking the user-provided chemical structure as the query, the top most similar compounds in terms of 3D similarity are returned with associated pharmacology annotations. ChemMapper is designed to provide versatile services in a variety of chemogenomics, drug repurposing, polypharmacology, novel bioactive compounds identification and scaffold hopping studies.
Software
SimG
-
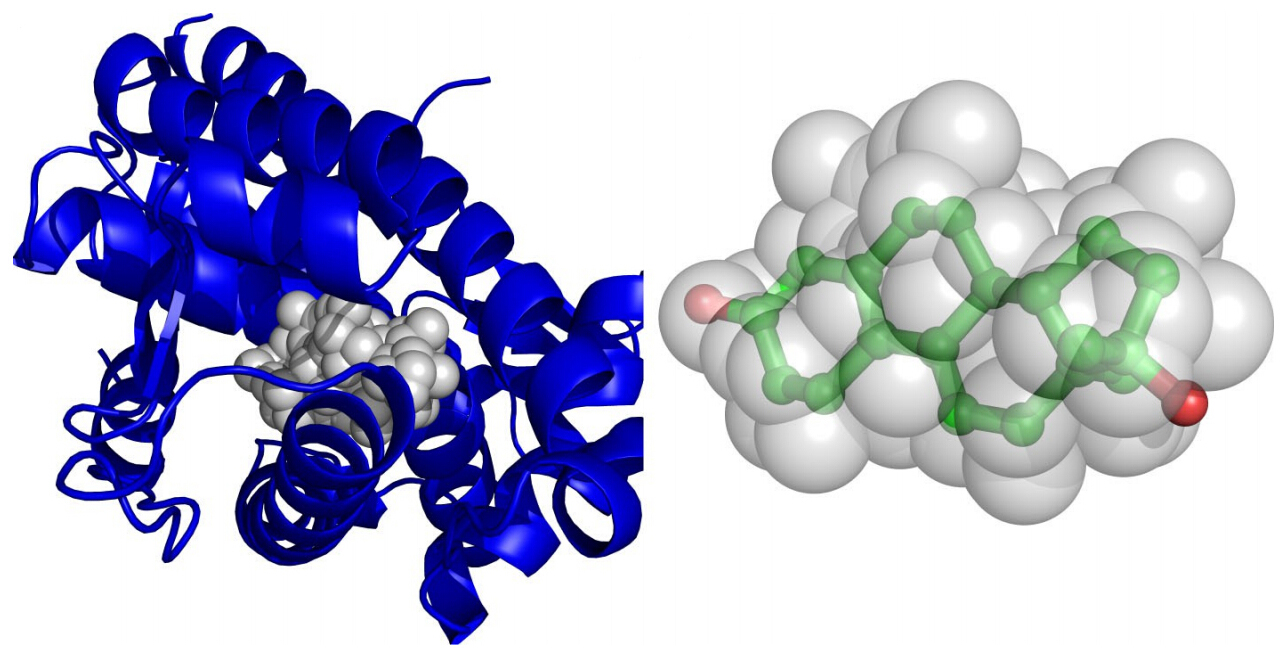 A molecular 3D similarity method named SimG is presented which applies a downhill simplex method to compare the shapes and chemical features of a small molecule and a binding pocket (or ligand) with Gaussian volume overlap in order to identify the active compounds. The basic idea is quite similar to ROCS that the molecular similarity is evaluated by performing a Gaussian volume based alignment, but the proposed method is featured in two aspects. First, both structure-based and ligand-based strategies are supported. Second, a downhill simplex searching algorithm was then carried out to effectively evaluate the shape and chemical feature similarity between database molecules and the query binding site (or ligand) which could be used as a criterion to retrieve active molecules.
A molecular 3D similarity method named SimG is presented which applies a downhill simplex method to compare the shapes and chemical features of a small molecule and a binding pocket (or ligand) with Gaussian volume overlap in order to identify the active compounds. The basic idea is quite similar to ROCS that the molecular similarity is evaluated by performing a Gaussian volume based alignment, but the proposed method is featured in two aspects. First, both structure-based and ligand-based strategies are supported. Second, a downhill simplex searching algorithm was then carried out to effectively evaluate the shape and chemical feature similarity between database molecules and the query binding site (or ligand) which could be used as a criterion to retrieve active molecules. MpsDockzn
-
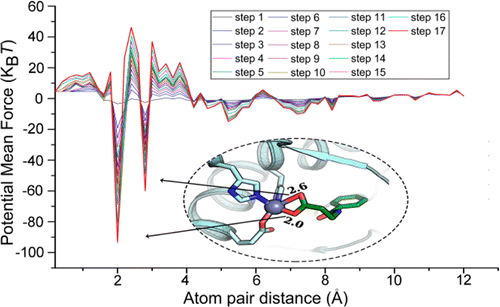 MpSDockZn is a zinc metalloprotein-specific molecular docking program, which employs NSGA II, a multi-objective optimization algorithm, to search the binding poses of a ligand to its metalloprotein receptor. Meanwhile, we designed a zinc metalloprotein-specific force field-based scoring function based on the recently reported zinc metalloprotein-specific non-bonded pairwise force field SLEF and developed a new zinc metalloprotein-specific knowledge-based scoring function based on the inverse Boltzmann law, nonlinear constrained optimization, and a designed dynamic sampling and iteration optimization strategy. These zinc metalloprotein-specific atom-pair potentials can reasonably and quantitatively describe the characteristics of different interactions between different atom-pairs, providing insights into the functions of these atom-pair potentials and highlighting important atom-pair interactions, such as coordination bonds, hydrogen bonds, and electrostatic interactions, thereby enhancing the ability to identify true-positive molecules and avoid the false-positive results. MpSDockZn is available at this site.
MpSDockZn is a zinc metalloprotein-specific molecular docking program, which employs NSGA II, a multi-objective optimization algorithm, to search the binding poses of a ligand to its metalloprotein receptor. Meanwhile, we designed a zinc metalloprotein-specific force field-based scoring function based on the recently reported zinc metalloprotein-specific non-bonded pairwise force field SLEF and developed a new zinc metalloprotein-specific knowledge-based scoring function based on the inverse Boltzmann law, nonlinear constrained optimization, and a designed dynamic sampling and iteration optimization strategy. These zinc metalloprotein-specific atom-pair potentials can reasonably and quantitatively describe the characteristics of different interactions between different atom-pairs, providing insights into the functions of these atom-pair potentials and highlighting important atom-pair interactions, such as coordination bonds, hydrogen bonds, and electrostatic interactions, thereby enhancing the ability to identify true-positive molecules and avoid the false-positive results. MpSDockZn is available at this site. PocketShape
-
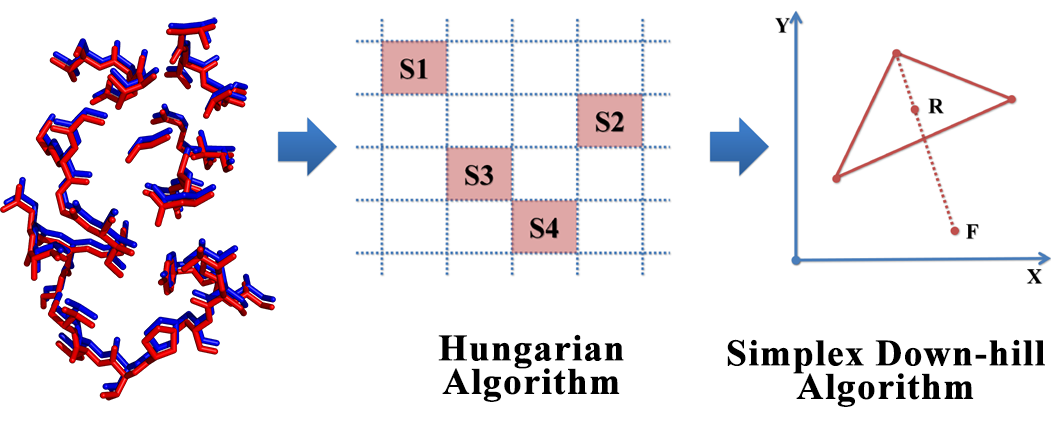 The interactions between proteins and other molecules are critical to a lot of biological processes, e.g. cellular signaling, regulation of cell cycles. Due to the fact that the functions of proteins are not necessarily correlated with protein sequence or fold, local feature analysis of proteins especially the pattern of binding sites may be another way to further understand protein functions and structures. In this study, a 3D structure based evaluation method for exploring the binding site similarity named PocketShape is presented. By means of structure-based alignment, PocketShape is capable of detecting common patterns or residue reservation between binding sites which are not required to possess continuous residues or to be homologous. Further possible application of PocketShape includes: prediction of a drug candidate’s off-target interactions, prediction of new target for known drugs, prediction of a protein’s function, protein classification. A protein profiling test carried out on protein kinase dataset demonstrates that PocketShape is capable of retrieving binding sites with common pattern.
The interactions between proteins and other molecules are critical to a lot of biological processes, e.g. cellular signaling, regulation of cell cycles. Due to the fact that the functions of proteins are not necessarily correlated with protein sequence or fold, local feature analysis of proteins especially the pattern of binding sites may be another way to further understand protein functions and structures. In this study, a 3D structure based evaluation method for exploring the binding site similarity named PocketShape is presented. By means of structure-based alignment, PocketShape is capable of detecting common patterns or residue reservation between binding sites which are not required to possess continuous residues or to be homologous. Further possible application of PocketShape includes: prediction of a drug candidate’s off-target interactions, prediction of new target for known drugs, prediction of a protein’s function, protein classification. A protein profiling test carried out on protein kinase dataset demonstrates that PocketShape is capable of retrieving binding sites with common pattern. Cyndi
-
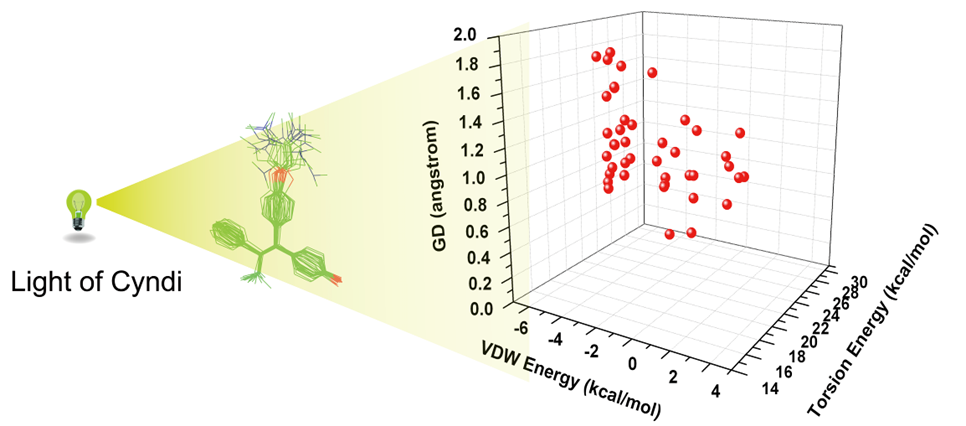 On the basis of MOEA algorithm, we present a new, highly efficient conformation generation method, Cyndi, and report the results of validation and performance studies comparing with other four methods. The results reveal that Cyndi is capable of generating geometrically diverse conformers and outperforms other four multiple conformer generators in the case of reproducing the bioactive conformations against 329 structures. The speed advantage indicates Cyndi is a powerful alternative method for extensive conformational sampling and large-scale conformer database preparation.
On the basis of MOEA algorithm, we present a new, highly efficient conformation generation method, Cyndi, and report the results of validation and performance studies comparing with other four methods. The results reveal that Cyndi is capable of generating geometrically diverse conformers and outperforms other four multiple conformer generators in the case of reproducing the bioactive conformations against 329 structures. The speed advantage indicates Cyndi is a powerful alternative method for extensive conformational sampling and large-scale conformer database preparation. eSHAFTS
-
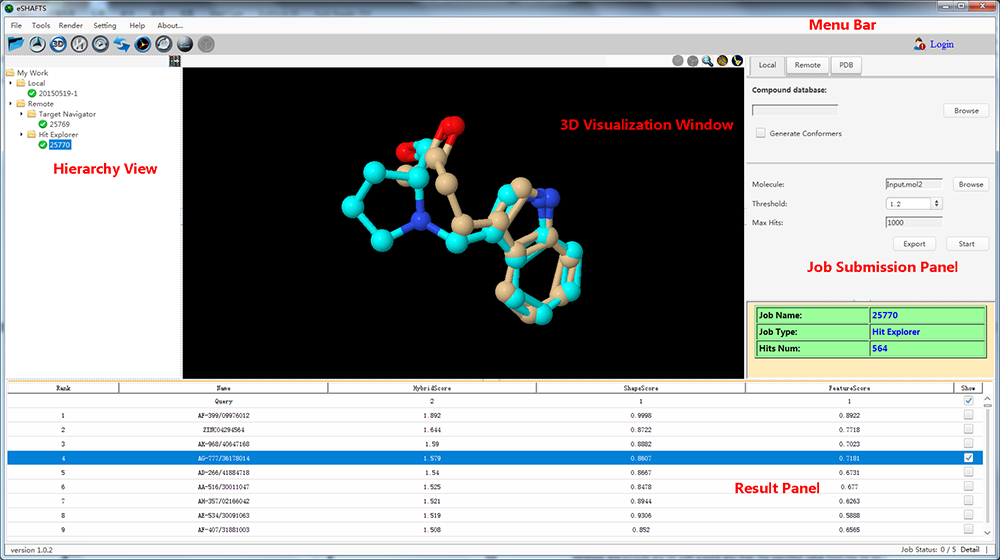 eSHAFTS, a user-friendly graphical working environment based on molecular 3D similarity method, has been developed. The overarching goal of eSHAFTS is to provide a computational platform that facilitates both experts and non-specialists to utilize common computational methodologies germane to drug discovery, such as drug repurposing, polypharmacology, hit compounds identification and scaffold hopping studies. To do so, eSHAFTS provides (1) a storage facility for chemical structures with bioactivities and associated target annotations (>350,000) and non-annotated compounds for virtual screening (>300,000), (2) an efficient combination of the SHAFTS, Cyndi, OpenBabel, JChemPaint and Jmol suites, (3) powerful user account management system, as well as data security and privacy protection mechanisms.
eSHAFTS, a user-friendly graphical working environment based on molecular 3D similarity method, has been developed. The overarching goal of eSHAFTS is to provide a computational platform that facilitates both experts and non-specialists to utilize common computational methodologies germane to drug discovery, such as drug repurposing, polypharmacology, hit compounds identification and scaffold hopping studies. To do so, eSHAFTS provides (1) a storage facility for chemical structures with bioactivities and associated target annotations (>350,000) and non-annotated compounds for virtual screening (>300,000), (2) an efficient combination of the SHAFTS, Cyndi, OpenBabel, JChemPaint and Jmol suites, (3) powerful user account management system, as well as data security and privacy protection mechanisms. iDNABinder
-
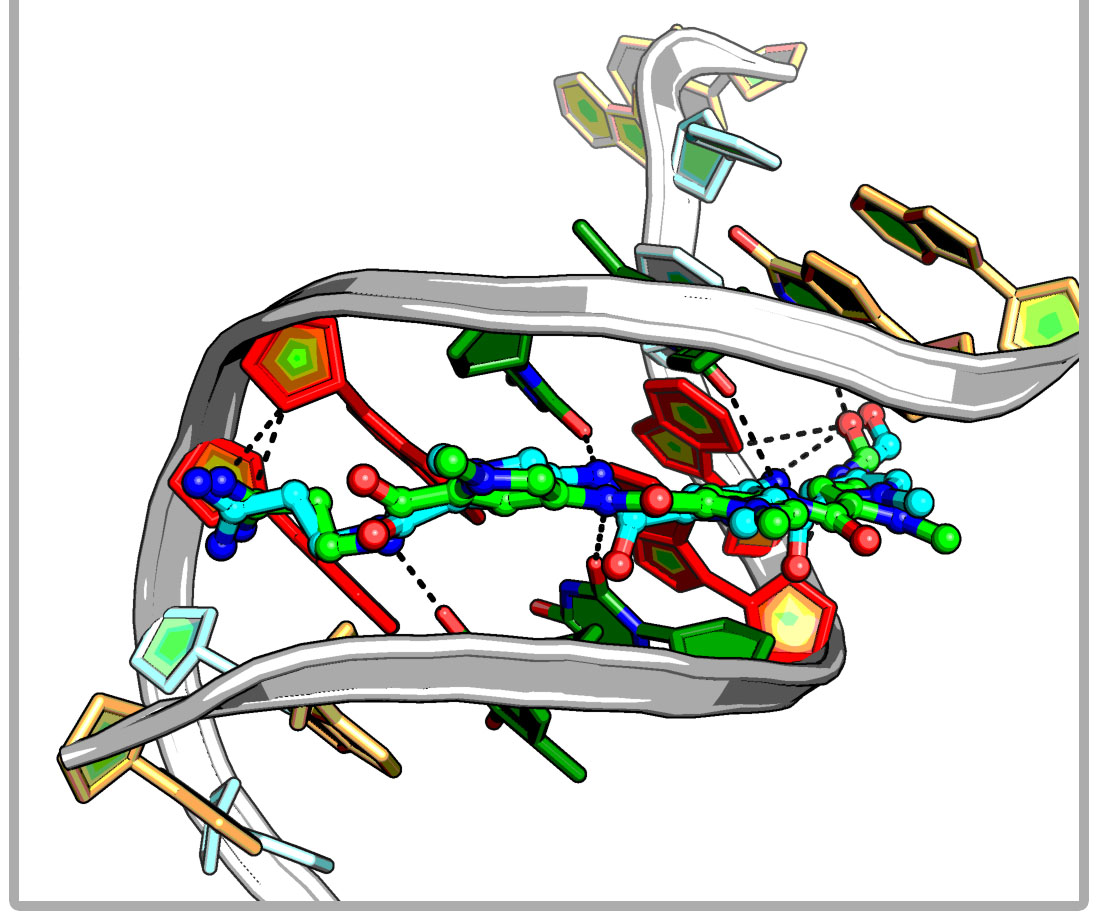 iDNABinder is an in-house specific molecular docking tool for DNA-ligand docking, whose scoring function comprises DNA-specific knowledge-based and force field-based scoring functions. It could be used to screen for small molecular docking or virtual screening against the specific DNA sequence and has been successfully applied to identifying potent natural modulator of AP-1, which selectively binds to a specific site (TRE 5`-TGACTCA-3`) of the AP-1 target DNA sequence and regulates AP-1-dependent gene transcription.
iDNABinder is an in-house specific molecular docking tool for DNA-ligand docking, whose scoring function comprises DNA-specific knowledge-based and force field-based scoring functions. It could be used to screen for small molecular docking or virtual screening against the specific DNA sequence and has been successfully applied to identifying potent natural modulator of AP-1, which selectively binds to a specific site (TRE 5`-TGACTCA-3`) of the AP-1 target DNA sequence and regulates AP-1-dependent gene transcription. SHAFTS
-
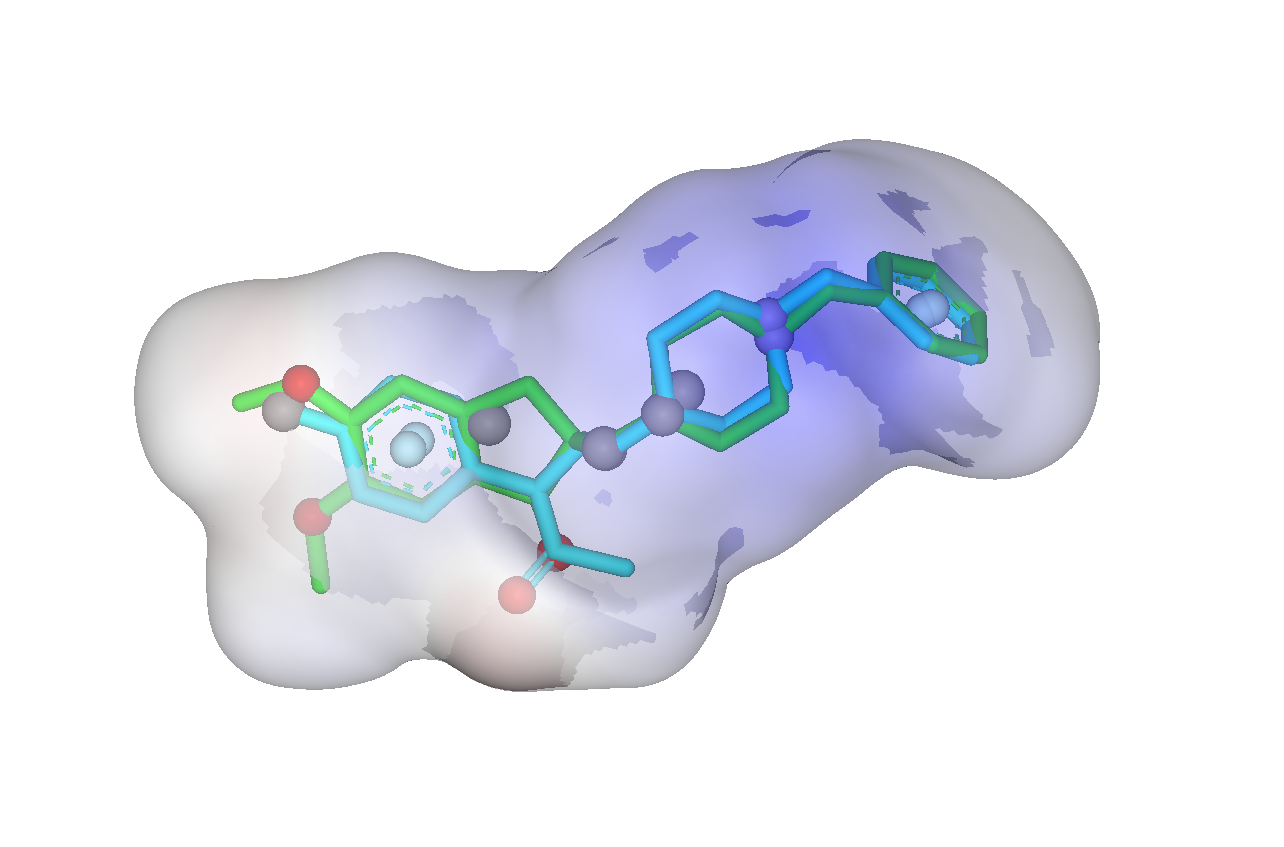 The SHAFTS method adopts hybrid similarity metric of molecular shape and colored (labeled) chemistry groups by pharmacophore features for 3D similarity calculation and ranking, which is designed to integrated the strength of both pharmacophore matching and volumetric overlay approaches. A feature triplet hashing method is used for fast molecular alignment poses enumeration, and the optimal superposition between the target and the query molecules can be prioritized by calculating corresponding “hybrid similarities”. SHAFTS is suitable for large-scale virtual screening with single or multiple bioactive compounds as the query“templates” regardless of whether corresponding experimentally determined conformations are available.
The SHAFTS method adopts hybrid similarity metric of molecular shape and colored (labeled) chemistry groups by pharmacophore features for 3D similarity calculation and ranking, which is designed to integrated the strength of both pharmacophore matching and volumetric overlay approaches. A feature triplet hashing method is used for fast molecular alignment poses enumeration, and the optimal superposition between the target and the query molecules can be prioritized by calculating corresponding “hybrid similarities”. SHAFTS is suitable for large-scale virtual screening with single or multiple bioactive compounds as the query“templates” regardless of whether corresponding experimentally determined conformations are available.
Contact us
Honglin Li's Lab
Shanghai Key Laboratory of New Drug Design
School of Pharmacy
East China University of Sci. & Tech.
Room 527, Building 18, 130 Meilong Road,
Shanghai, 200237, P. R. China
Tel: (86) 21 6425 0213 Prof. Honglin Li
hlli@ecust.edu.cn
Copyright © 2020 Prof. HongLin Li's Group, School of Pharmacy, East China University of Science & Technology · All Right Reserved.